
A CRISPR–Cas9-based therapeutic from Editas is poised to enter clinical trials for treating blindness
Ingrijpen in het DNA van de mens: doen of laten?
Mogelijkheden om het DNA van menselijke embryo’s te veranderen zijn in een stroomversnelling geraakt. Met het aanbrengen van wijzigingen in het embryonale DNA kunnen erfelijke ziekten bij het nageslacht worden voorkomen. Om de ontwikkelingen in goede banen te leiden is een geactualiseerd juridisch-ethisch beoordelingskader onmisbaar. Overheid, wetenschap en samenleving zullen bovendien samen moeten bepalen wie wanneer waarover besluit en op basis van welke argumenten. Dit schrijven de COGEM en de Gezondheidsraad in een gezamenlijk signalement aan de minister van VWS, de staatssecretaris van IenM en de Tweede Kamer. De Gezondheidsraad verbindt daaraan het advies de Embryowet aan te passen.
Recent zijn technieken ontwikkeld waarmee sneller en efficiënter dan voorheen zogeheten kiembaanmodificatie mogelijk wordt: het gericht veranderen van het DNA in embryo’s, waardoor het ontstane individu en diens nageslacht deze verandering doorgeven. Deze nieuwe mogelijkheden bieden perspectief voor de behandeling en preventie van erfelijke aandoeningen. Er zijn echter ook nog onzekerheden over de effectiviteit en veiligheid van kiembaanmodificatie, op zowel de korte als de lange termijn.
Met deze technisch-wetenschappelijke ontwikkelingen doen zich tal van vragen en dilemma’s voor. Wat zijn de juridische, ethische en maatschappelijke implicaties van kiembaanmodificatie? Waar komen de huidige juridische en ethische kaders onder druk te staan? Hoe af te wegen welke vormen van onderzoek en klinische toepassing verantwoord zijn? De COGEM en de Gezondheidsraad achten het van groot belang dat overheid, wetenschap, medische professionals en samenleving gezamenlijk bepalen hoe de besluitvorming over kiembaanmodificatie verder vorm krijgt. Dit signalement biedt daarvoor een handreiking.
Voor nader onderzoek naar de mogelijkheden van kiembaanmodificatie met de nieuwe technieken zijn zogenoemde kweekembryo’s nodig. Op grond van de huidige Embryowet is slechts onderzoek toegestaan met embryo’s die overblijven na vruchtbaarheidsbehandelingen. Het speciaal voor onderzoeksdoeleinden kweken van embryo’s wordt gezien als een te grote inbreuk op het respect voor leven. Vorig jaar heeft de regering echter een verruiming van de Embryowet aangekondigd. In dit verband adviseert de Gezondheidsraad de minister van VWS om het verbod op onderzoek met kweekembryo’s op te heffen. Als dergelijk onderzoek gericht wordt op het voorkomen van ernstige aandoeningen en aan strikte voorwaarden wordt gebonden, is het volgens de raad niet in strijd met de menselijke waardigheid. Wel zou daarbij een maatschappelijke dialoog moeten worden gevoerd, zowel met het werkveld als met het bredere publiek.
Het rapport (Ingrijpen in het DNA van de mens: Morele en maatschappelijke implicaties van kiembaanmodificatie) is te downloaden van de websites www.gezondheidsraad.nl en www.cogem.net. Nadere inlichtingen verstrekken Eert Schoten (Gezondheidsraad), tel. 06 462 36 998, e-mail: ej.schoten@gr.nl en Frank van der Wilk (COGEM), tel. 030-2742777, e-mail: frank.vanderwilk@cogem.net
Source: Nieuws | Cogem NL
Consensus statement of European Societies of Gene and Cell Therapy on the reported birth of genome-edited babies in China
European Society of Gene and Cell Therapy (ESGCT), British Society for Gene and Cell Therapy (BSGCT), Deutsche Gesellschaft für Gentherapie (DG-GT), Finnish Society of Gene Therapy (FSGT), Hellenic Society of Gene Therapy and Regenerative Medicine (HSGTRM), Netherlands Society for Gene & Cell Therapy (NVGCT), Sociedad Española de Terapia Génica y Celular (SETGyC), Société Française de Thérapie Cellulaire et Génique (SFTCG)

Somatic gene therapy has been developed to treat inherited and acquired diseases, for which there are currently no or only limited other treatment options. Genetic modifications are restricted to cells of the body (somatic cells), but exclude germline cells to avoid transmission of genetic changes to the offspring. Our societies have supported the development of innovative approaches of somatic gene and cell therapies for more than 25 years. While we acknowledge the necessity of fundamental research, our societies agree that based on current knowledge and experience, germline genome editing is irresponsible and ethically not justifiable.
Documents posted to the Chinese clinical trial registry and articles published by various news outlets suggest that Dr. Jiankui He (Southern University of Science and Technology, Shenzhen) used CRISPR/Cas9 designer nuclease technology to alter the genome of human embryos prior to uterus implantation. CRISPR/Cas9 is a state-of-the art technology in molecular medicine for research purposes and has also been developed as a tool for gene therapy. The technology can be described as molecular scissors that allow to modify the cellular genome at pre-defined, so called target sites, and thus opens the possibility to repair a genetic defect. Nonetheless, off-target activity, i.e. cutting at non-intended sites in the human genome, cannot completely be excluded. Off-target activity can generate mutations with unknown effects not only on the subjects’ health but also on that of their progeny.
Dr. He has given an exclusive interview and posted a video on YouTube describing his research, a very unusual way to present scientific work. He has declared that healthy twin girls were born after genome editing of embryos generated by in-vitro fertilisation (IVF). He claims that in at least one of the twin girls he successfully inactivated the CCR5 gene thereby protecting the girl from infection with human immunodeficiency virus (HIV). Besides the potential side effects and the huge ethical implications, even the scientific reasoning for his approach is very questionable for several reasons: (i) The reported CCR5 knockout would not, as stated, result in complete protection from HIV, (ii) The CCR5 knockout itself could have potentially serious functional consequences, as for example the already established higher susceptibility to viruses other than HIV, (iii) Given the current efforts to decrease the rate of new HIV infections and to develop effective vaccines, in addition to self-protection, the foreseeable risk of HIV infection is low.
To the best of our knowledge, the claims by Dr. He has so far not been confirmed by an independent and scientifically sound source. The research has not been peer-reviewed, but was presented at the Second International Summit on Human Genome Editing, which is taking place 27-29 November 2018 in Hong Kong. According to Chinese government and Shenzhen University officials cited in the media, Dr. He had not obtained permission by the competent authorities, including the responsible Ethical committee.
Gene and cell therapies have huge therapeutic potential, and genome editing is currently emerging as a promising and powerful tool for repairing genetic defects. As with any treatment, however, there are potential risks associated with genome editing that need careful investigation before these new treatments can be used in patients. Besides the necessary safety studies, application to germline modification, in our view, requires broad societal discussion of the ethical implications. The Second International Summit on Human Genome Editing taking place this week is one of the many international efforts to find an international consensus. Importantly, there has been a general agreement at the first Summit: “It would be irresponsible to proceed with any clinical use of germline editing unless and until (i) the relevant safety and efficacy issues have been resolved, based on appropriate understanding and balancing of risks, potential benefits, and alternatives, and (ii) there is broad societal consensus about the appropriateness of the proposed application.” The ESGCT and the named European Societies completely support this view and condemn Dr. He’s experiments as risky, designed for enhancement rather than therapy, and therefore ethically inacceptable. We recognize the power of this technology for treatment of human diseases, but urge caution until further research and experience with genome editing, public consultation and legislation is consensually established. Lawmakers and the public should have a say about the circumstances, under which the use of these cutting-edge technologies is permissible. While a number of rigorously tested gene and cell therapies are beginning to realise their potential in the clinic, it is important that clinical studies continue to meet international standards, to ensure they are safe, and that research is conducted transparently and responsibly. Irresponsible and unethical acceleration of the use of these technique in the clinic prior to legislative debates is more likely to hinder than accelerate the progress of these promising techniques.
Signed on behalf of the Societies,
Hildegard Büning (ESGCT), Uta Griesenbach (BSGCT), Boris Fehse (DG-GT), Seppo Ylä-Herttuala (FSGT), Nicholas P. Anagnou (HSGTRM), Victor van Beusechem (NVGCT), Angel Raya (SETGyC), Els Verhoeyen (SFTCG)
Chinese scientists are creating CRISPR babies
A daring effort is under way to create the first children whose DNA has been tailored using gene editing.
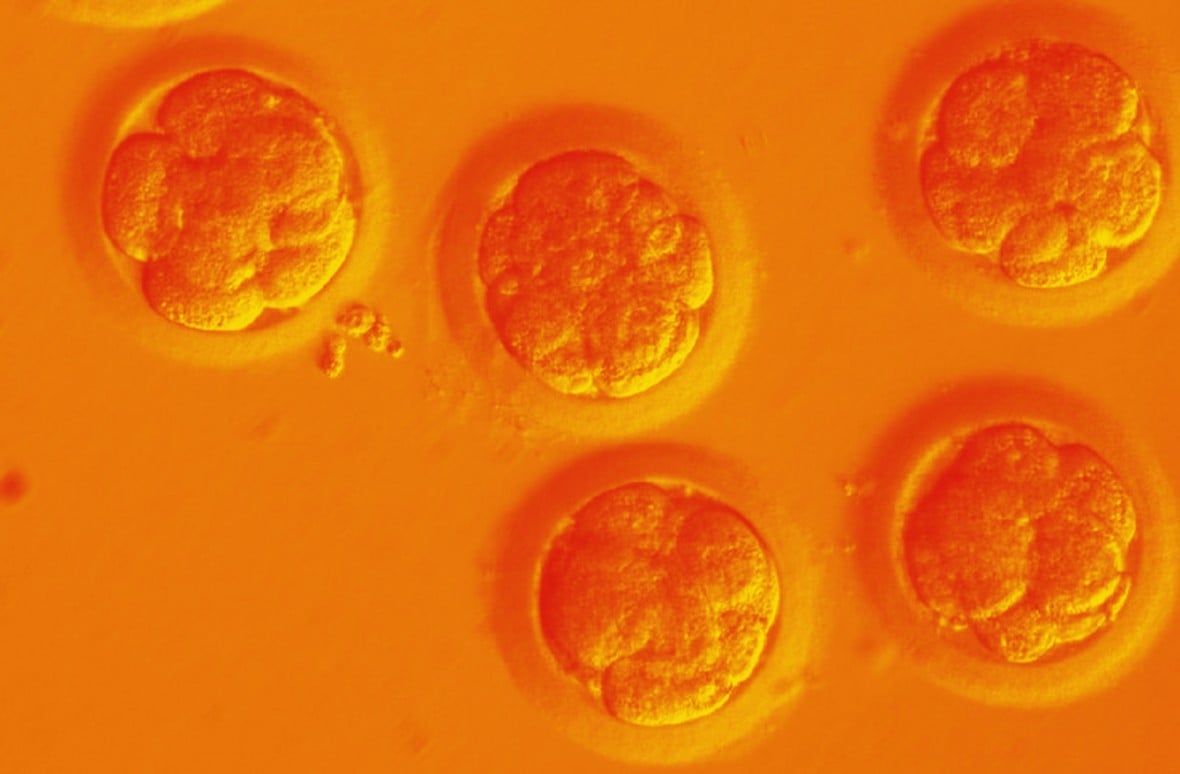
When Chinese researchers first edited the genes of a human embryo in a lab dish in 2015, it sparked global outcry and pleas from scientists not to make a baby using the technology, at least for the present.
It was the invention of a powerful gene-editing tool, CRISPR, which is cheap and easy to deploy, that made the birth of humans genetically modified in an in vitro fertilization (IVF) center a theoretical possibility.
Now, it appears it may already be happening.
According to Chinese medical documents posted online this month (here and here), a team at the Southern University of Science and Technology, in Shenzhen, has been recruiting couples in an effort to create the first gene-edited babies. They planned to eliminate a gene called CCR5 in hopes of rendering the offspring resistant to HIV, smallpox, and cholera.

SOUTHERN UNIVERSITY OF SCIENCE AND TECHNOLOGY
The clinical trial documents describe a study in which CRISPR is employed to modify human embryos before they are transferred into women’s uteruses.
The scientist behind the effort, He Jiankui, did not reply to a list of questions about whether the undertaking had produced a live birth. Reached by telephone, he declined to comment.
However, data submitted as part of the trial listing shows that genetic tests have been carried out on fetuses as late as 24 weeks, or six months. It’s not known if those pregnancies were terminated, carried to term, or are ongoing.
[After this story was published, the Associated Press reported that according to He, one couple in the trial gave birth to twin girls this month, though the agency wasn’t able to confirm his claim independently. He also released a promotional video about his project.]The birth of the first genetically tailored humans would be a stunning medical achievement, for both He and China. But it will prove controversial, too. Where some see a new form of medicine that eliminates genetic disease, others see a slippery slope to enhancements, designer babies, and a new form of eugenics. The step toward genetically tailored humans was undertaken in secrecy and with the clear ambition of a stunning medical first.
“In this ever more competitive global pursuit of applications for gene editing, we hope to be a stand-out,” He and his team wrote in an ethics statement they submitted last year. They predicted their innovation “will surpass” the invention of in vitro fertilization, whose developer was awarded a Nobel Prize in 2010.
Gene-editing summit
The claim that China has already made genetically altered humans comes just as the world’s leading experts are jetting into Hong Kong for the Second International Summit on Human Genome Editing.
The purpose of the international meeting is to help determine whether humans should begin to genetically modify themselves, and if so, how. That purpose now appears to have been preempted by the actions of He, an elite biologist recruited back to China from the US as part of its “Thousand Talents Plan.”
The technology is ethically charged because changes to an embryo would be inherited by future generations and could eventually affect the entire gene pool. “We have never done anything that will change the genes of the human race, and we have never done anything that will have effects that will go on through the generations,” David Baltimore, a biologist and former president of the California Institute of Technology, who chairs the international summit proceedings, said in a pre-recorded message ahead of the event, which begins Tuesday, November 27.
It appears the organizers of the summit were also kept in the dark about He’s plans.
Regret and concern
The genetic editing of a speck-size human embryo carries significant risks, including the risks of introducing unwanted mutations or yielding a baby whose body is composed of some edited and some unedited cells. Data on the Chinese trial site indicate that one of the fetuses is a “mosaic” of cells that had been edited in different ways.
A gene-editing scientist, Fyodor Urnov, associate director of the Altius Institute for Biomedical Sciences, a nonprofit in Seattle, reviewed the Chinese documents and said that, while incomplete, they do show that “this effort aims to produce a human” with altered genes.
Urnov called the undertaking cause for “regret and concern over the fact that gene editing—a powerful and useful technique—was put to use in a setting where it was unnecessary.” Indeed, studies are already under way to edit the same gene in the bodies of adults with HIV. “It is a hard-to-explain foray into human germ-line genetic engineering that may overshadow in the mind of the public a decade of progress in gene editing of adults and children to treat existing disease,” he says.
Big project
In a scientific presentation in 2017 at Cold Spring Harbor Laboratory, which is posted to YouTube, He described a very large series of preliminary experiments on mice, monkeys, and more than 300 human embryos. One risk of CRISPR is that it can introduce accidental or “off target” mutations. But He claimed he found few or no unwanted changes in the test embryos.
He is also the chairman and founder of a DNA sequencing company called Direct Genomics. A new breed of biotech companies could ultimately reap a windfall should the new methods of conferring health benefits on children be widely employed.

THE NATIONAL ACADEMIES OF SCIENCE, ENGINEERING AND MEDICINE
According to the clinical trial plan, genetic measurements would be carried out on embryos and would continue during pregnancy to check on the status of the fetuses. During his 2017 presentation, He acknowledged that if the first CRISPR baby were unhealthy, it could prove a disaster.
“We should do this slow and cautious, since a single case of failure could kill the whole field,” he said.
A listing describing the study was posted in November, but other trial documents are dated as early as March of 2017. That was only a month after the National Academy of Sciences in the US gave guarded supportfor gene-edited babies, although only if they could be created safely and under strict oversight.
Currently, using a genetically engineered embryo to establish a pregnancy would be illegal in much of Europe and prohibited in the United States. It is also prohibited in China under a 2003 ministerial guidance to IVF clinics. It is not clear if He got special permission or disregarded the guidance, which may not have the force of law.
Public opinion
In recent weeks, He has begun an active outreach campaign, speaking to ethics advisors, commissioning an opinion poll in China, and hiring an American public-relations professional, Ryan Ferrell.
“My sense is that the groundwork for future self-justification is getting laid,” says Benjamin Hurlbut, a bioethicist from Arizona State University who will attend the Hong Kong summit.
The new opinion poll, which was carried out by Sun Yat-Sen University, found wide support for gene editing among the sampled 4,700 Chinese, including a group of respondents who were HIV positive. More than 60% favored legalizing edited children if the objective was to treat or prevent disease. (Polls by the Pew Research Center have found similar levels support in the US for gene editing.)
He’s choice to edit the gene called CCR5 could prove controversial as well. People without working copies of the gene are believed to be immune or highly resistant to infection by HIV. In order to mimic the same result in embryos, however, He’s team has been using CRISPR to mutate otherwise normal embryos to damage the CCR5 gene.
The attempt to create children protected from HIV also falls into an ethical gray zone between treatment and enhancement. That is because the procedure does not appear to cure any disease or disorder in the embryo, but instead attempts to create a health advantage, much as a vaccine protects against chicken pox.
For the HIV study, doctors and AIDS groups recruited Chinese couples in which the man was HIV positive. The infection has been a growing problem in China.
So far, experts have mostly agreed that gene editing shouldn’t be used to make “designer babies” whose physical looks or personality has been changed.
He appeared to anticipate the concerns his study could provoke. “I support gene editing for the treatment and prevention of disease,” He posted in November to the social media site WeChat, “but not for enhancement or improving I.Q., which is not beneficial to society.”
Still, removing the CCR5 gene to create HIV resistance may not present a particularly strong reason to alter a baby’s heredity. There are easier, less expensive ways to prevent HIV infection. Also, editing embryos during an IVF procedure would be costly, high-tech, and likely to remain inaccessible in many poor regions of the world where HIV is rampant.
A person who knows He said his scientific ambitions appear to be in line with prevailing social attitudes in China, including the idea that the larger communal good transcends individual ethics and even international guidelines.
Behind the Chinese trial also lies some bold thinking about how evolution can be shaped by science. While the natural mutation that disables CCR5 is relatively common in parts of Northern Europe, it is not found in China. The distribution of the genetic trait around the world—in some populations but not in others—highlights how genetic engineering might be used to pick the most useful inventions discovered by evolution over the eons in different locations and bring them together in tomorrow’s children.
Such thinking could, in the future, yield people who have only the luckiest genes and never suffer Alzheimer’s, heart disease, or certain infections.
The text of an academic website that He maintains shows that he sees the technology in the same historic, and transformative, terms. “For billions of years, life progressed according to Darwin’s theory of evolution,” it states. More recently, industrialization has changed the environment in radical ways posing a “great challenge” that humanity can meet with “powerful tools to control evolution.”
It concludes: “By correcting the disease genes … we human[s] can better live in the fast-changing environment.”
Note: This story was updated after publication to include claims by He Jiankui that the trial had produced live births.
Cel- en gentherapie in opkomst maar toepassing kent belemmeringen – ZonMw
De komende jaren komen voor verschillende vormen van kanker en andere ernstige ziekten behandelingen met celtherapie en gentherapie beschikbaar.
Met deze zogenoemde advanced therapy medicinal products (ATMP’s) kunnen bijvoorbeeld afweercellen van de patiënt worden bewerkt om een tumor te herkennen en uit te schakelen. Een andere veelbelovende ontwikkeling is gentherapie voor zeldzame aandoeningen als hemofilie B. Deze ziekte is met gentherapie mogelijk te genezen. Dit blijkt uit een verkenning van de ontwikkelingen van ATMP’s die het RIVM in opdracht van het ministerie van VWS heeft uitgevoerd.
De meeste ATMP’s zijn bedoeld om ernstige, vaak zeldzame aandoeningen te behandelen. Ze kunnen levensverlengend, soms zelfs genezend zijn, of de kwaliteit van leven sterk verbeteren. Ondanks de ontwikkelingen van de afgelopen 20 jaren zijn de hoge verwachtingen nog niet uitgekomen. Wel blijft de techniek hoopgevend en worden er tussen 5 en 10 jaar nieuwe producten verwacht.
Eigenschappen toegevoegd
In algemene zin wordt er bij ATMP’s een (bijvoorbeeld ontbrekende) eigenschap aan cellen toegevoegd die bepaalde processen in het lichaam in gang zet. Zo kunnen bijvoorbeeld kankercellen worden uitgeschakeld of een stollingsfactor worden gemaakt. In Nederland wordt veel onderzoek met ATMP’s bij patiënten gedaan en stijgt het aantal studies met ATMP’s die in een vergevorderde ontwikkelingsfase zijn.
Belemmeringen
De onderzoekers constateren belemmeringen die een succesvolle behandeling met ATMP’s vooralsnog in de weg staan. Zo zijn ATMP’s moeilijk in te passen in de huidige wetgeving voor markttoelating van geneesmiddelen. Andere belemmerende factoren zijn financiering (de ontwikkeling van ATMP’s is kostbaar)en de maatschappelijke druk om ATMP’s al beschikbaar te maken voor patiënten zonder dat het werkingsmechanisme goed bekend is.
Meer informatie
- u kunt in het ATMP rapport 2017 meer over toekomstverwachtingen van ATMP’s lezen
Source: Cel- en gentherapie in opkomst maar toepassing kent belemmeringen – ZonMw